
DeepPolisher — это новый инструмент глубокого обучения, который значительно повышает точность сборки генома за счет точной коррекции ошибок на уровне оснований, и недавно он сыграл ключевую роль в улучшении эталонного пангенома человека.
Быстрые ссылки
- Бумага
- Репозиторий кода
- Делиться
Ключ к пониманию наследственности, болезней и эволюции лежит в геноме, который закодирован нуклеотидами (то есть основаниями A, T, G и C). Секвенаторы ДНК могут считывать эти нуклеотиды, но сделать это точно и в больших масштабах сложно из-за очень малого размера пар оснований. Однако, чтобы раскрыть тайны, скрытые в геноме, мы должны иметь возможность собрать эталонный геном, максимально приближенный к идеальному.
Ошибки при сборке генома могут ограничивать методы, используемые для идентификации генов и белков, и могут привести к тому, что последующие диагностические процессы пропустят варианты, вызывающие заболевания. При сборке генома один и тот же геном секвенируется много раз, что позволяет итеративно исправлять ошибки. Тем не менее, учитывая, что человеческий геном состоит из 3 миллиардов нуклеотидов, даже небольшая частота ошибок может означать большое общее количество ошибок и ограничивать полезность полученного генома.
Стремясь постоянно улучшать ресурсы для сборки генома, мы представляем DeepPolisher — метод сборки генома с открытым исходным кодом, разработанный нами в сотрудничестве с Институтом геномики Калифорнийского университета в Санта-Крузе. В нашей недавней статье «Высокоточная доработка сборки с помощью DeepPolisher», опубликованной в журнале Genome Research , мы описываем, как этот конвейер расширяет существующие методы для повышения точности сборки генома. DeepPolisher снижает количество ошибок в сборке на 50% и количество ошибок вставки или удаления («инделов») на 70%. Это особенно важно, поскольку ошибки инделов мешают идентификации генов.
Фон
Хотя существует несколько способов измерения ДНК, большинство из них обычно включают в себя фиксацию процесса копирования ДНК. Один из методов заключается в присоединении молекул-меток разных цветов к раздельным нуклеотидам-строительным блокам и наблюдении за процессом добавления каждого из них к копируемой молекуле ДНК. Механизм копирования ДНК всегда копирует нить в определенной ориентации, поэтому, хотя информация избыточно кодируется на обеих нитях, считываются только нуклеотиды с одной нити за раз. Идентификация нуклеотидов требует детекторов, способных различать отдельные молекулы, что ограничивает точность измерений.
Одна из прорывных технологий, разработанная компанией Illumina для масштабирования этого метода, копирует одну молекулу секвенируемой ДНК в кластер идентичных копий. Затем она отслеживает синхронное копирование кластера, тем самым увеличивая сигнал для каждой нуклеотидной основы. Однако, поскольку невозможно гарантировать идеальное синхронное копирование кластера, он может рассинхронизироваться, в результате чего сигналы от разных оснований смешиваются, что ограничивает длину ДНК, измеряемой этим методом, несколькими сотнями нуклеотидов.
Хотя эти последовательности (называемые «ридами») короткие, они все же полезны для анализа. Сравнивая их с эталонным геномом, то есть с существующей картой генома вида, который необходимо секвенировать, можно сопоставить многие короткие риды с этим эталоном, тем самым создавая более полный геном исследуемого организма. Затем его можно сравнить с эталоном, чтобы лучше понять, как изменяется геном исследуемого организма.

Геном человека состоит из двух нитей, которые избыточно кодируют информацию ( слева ), организованных в хромосомы, при этом от каждого родителя наследуется одна полная копия ( справа ). ( Изображения предоставлены NHGRI )
Даже с усовершенствованными технологиями секвенирования сохраняется ряд проблем. Во-первых, этот метод основан на наличии надежного референсного генома, создание которого само по себе чрезвычайно сложно. Даже при наличии такого референса некоторые участки генома больше похожи на другие, что затрудняет их уверенное сопоставление с референсом.
Для решения этих проблем ученые разработали процессы, позволяющие секвенировать отдельные молекулы, что дает возможность считывать десятки тысяч нуклеотидов. Первоначально этот процесс имел неприемлемо высокий уровень ошибок (~10%). Эта проблема была решена, когда компания Pacific Biosciences разработала способ секвенирования одной и той же молекулы в несколько этапов, снизив уровень ошибок до 1%, аналогично методам секвенирования коротких фрагментов. Google и Pacific Biosciences совместно продемонстрировали это на примере генома человека.
Наша команда пошла дальше, разработав DeepConsensus, который использует преобразователь последовательностей для более точного построения правильной последовательности из исходных оснований, подверженных ошибкам. Сегодня Pacific Biosciences использует DeepConsensus на своих секвенаторах длинных прочтений, чтобы снизить частоту ошибок до менее чем 0,1%. Хотя эта частота ошибок значительно лучше, чем у предыдущих передовых методов, достижение точности, необходимой для построения нового, практически идеального эталонного генома, требует объединения прочтений последовательностей из нескольких молекул ДНК одного и того же индивидуума для дальнейшей коррекции оставшихся ошибок.
ДипПолишер
Здесь на помощь приходит DeepPolisher. Адаптированный из DeepConsensus, DeepPolisher использует архитектуру Transformer, обученную на геноме линии клеток человека, предоставленной проекту Personal Genomes Project. Этот эталонный геном был исчерпывающе охарактеризован NIST и NHGRI и секвенирован с использованием множества различных технологий. Его полнота оценивается примерно в 100%, а точность — в 99,99999%. Это соответствует примерно 300–1000 общим ошибкам на 6 миллиардах нуклеотидов в геноме (две копии эталонного генома из 3 миллиардов нуклеотидов, унаследованные от каждого родителя).
Проведя секвенирование и сборку генома с помощью PacBio, мы можем выявить оставшиеся ошибки, а затем обучить модели их исправлять. Для обучения модель принимает во внимание секвенированные основания, их качество и то, насколько однозначно они соответствуют заданной части эталонной сборки. Во время обучения мы используем только хромосомы 1–19. Мы исключаем хромосомы 20–22, используя результаты на хромосомах 21 и 22 для выбора модели, и сообщаем о точности, используя хромосому 20.

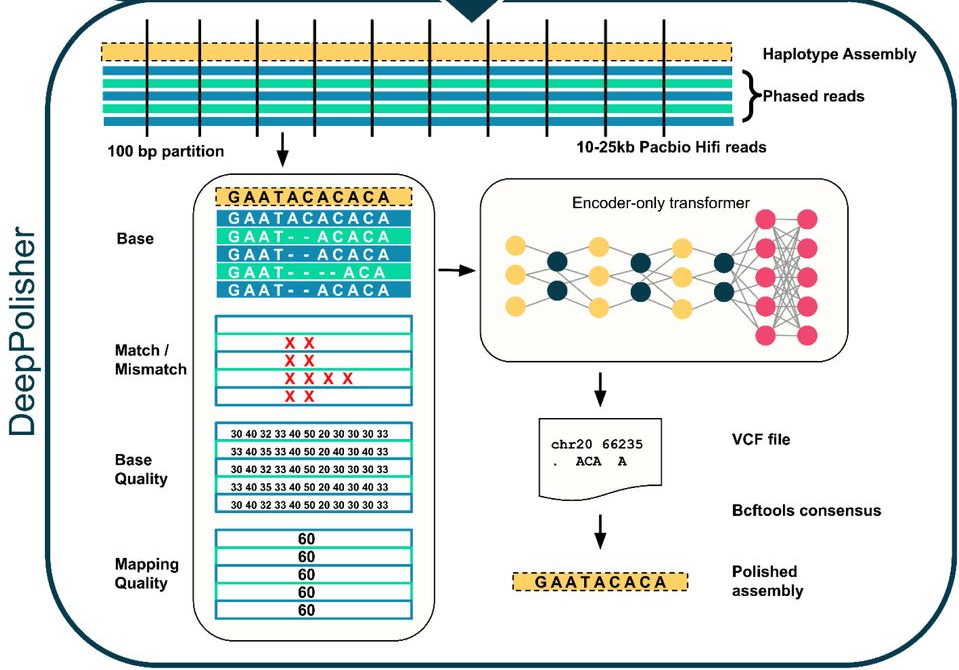
Архитектура DeepPolisher. Последовательности прочтений классифицируются по происхождению (так называемое «фазирование») и выравниваются по черновому варианту сборки генома. Входные каналы: информация о основаниях, сообщаемое секвенатором качество, качество картирования (способность однозначно размещать прочтения в сборке) и аннотации несовпадающих оснований. Эти данные передаются в преобразователь, работающий только с кодировщиком, который классифицирует ошибки в сборке и предлагает исправление, используемое для корректировки сборки.
Производительность
DeepPolisher уменьшает количество ошибок в сборке генома примерно вдвое, что в значительной степени обусловлено сокращением ошибок вставки-удаления («инделов»), которые уменьшаются более чем на 70 процентов. Сокращение этих типов ошибок особенно важно, поскольку вставленные или удаленные основания могут сдвигать рамку считывания гена, из-за чего программы аннотирования могут игнорировать этот ген при маркировке генома и скрывать его от отчетов в клиническом анализе или разработке лекарств.
Мы количественно оцениваем качество генома с помощью «Q-балла», который представляет собой десятичный логарифм вероятности ошибки в определенной позиции генома. Показатель Q30 означает 99,9% вероятности правильности, а Q60 — 99,9999% вероятности правильности основания. Для оценки улучшения DeepPolisher мы использовали данные секвенирования, применяемые для сборки новых геномов для Консорциума эталонных геномов человека (HPRC). Мы искали потенциальные ошибки в сборке, пытаясь выявить комбинации нуклеотидов, которые не встречаются при других методах секвенирования того же образца с использованием других технологий секвенирования. Проведя этот анализ в тех частях генома, для которых другой метод секвенирования не имеет систематических ошибок (доверительный регион), мы можем показать улучшение сборки в среднем с Q66,7 до Q70,1. Мы также демонстрируем улучшение для каждого отдельного образца, подвергнутого оценке.

Качество сборки до и после полировки для 180 образцов. Для каждого образца геном разделен по родительскому происхождению (копия генома, переданная отцом или матерью), обозначенному как гаплотип (Hap) 1 или 2, и оценено качество этих гаплотипов.
Развертывание
DeepPolisher уже используется для улучшения геномных ресурсов для научного сообщества. В мае HPRC объявил о выпуске второго релиза данных, который включал секвенированные сборки геномов 232 человек, что в пять раз больше, чем в первом релизе. Данные во втором релизе прошли дополнительную обработку с помощью DeepPolisher, что вдвое снизило количество ошибок в виде отдельных нуклеотидов и инсерций/делеций, в результате чего частота ошибок составила менее одной ошибки на полмиллиона собранных оснований.
Предоставляя DeepPolisher в качестве инструмента с открытым исходным кодом, мы стремимся сделать методы доступными для широкого сообщества. Работая с Консорциумом эталонных данных человеческого пангенома, мы помогаем ученым более точно диагностировать генетические заболевания у людей всех этнических групп.
Благодарности
В этом посте в блоге демонстрируется вклад Google в разработку DeepPolisher для повышения качества сборки геномов. Интеграция DeepPolisher в более широкий контекст создания высокоточных пангеномных эталонов включает в себя вклад почти 195 авторов из 68 различных организаций. Мы благодарим исследовательские группы Института геномики Калифорнийского университета в Санта-Круз (UCSC Genomics Institute, GI) под руководством профессора Бенедикта Патена и профессора Карен Мига за помощь в первичном анализе и определении направлений разработки DeepPolisher. Мы выражаем признательность Мире Масторас и Мобину Асри за руководство основным анализом и интеграцией DeepPolisher в конвейер генерации пангеномов. Мы благодарим технических специалистов Google: Пи-Чуан Чанга, Даниэля Э. Кука, Алексея Колесникова, Лукаса Брамбринка и Марию Наттестад. Мы благодарим Лиззи Дорфман, Дейла Вебстера и Кэтрин Чоу за стратегическое руководство, а также Моник Бруйетт за помощь в написании статьи.
Источник: research.google